The Basics of Cystic Fibrosis
Reviewed by: HU Medical Review Board | Last reviewed: September 2019 | Last updated: June 2023
Cystic fibrosis (CF) is a genetically inherited disease that results in persistent lung infections and gastrointestinal issues that gradually damage many organs. It affects the lungs the most, but also the digestive system, including the pancreas, liver, intestines, and kidneys. CF may also impact the ears, nose and sinuses, sex organs, and the bones and joints.1,2
Cystic fibrosis is usually diagnosed soon after birth, though in milder cases the person may not be diagnosed until adulthood.3 The Cystic Fibrosis Foundation estimates that more than 30,000 children and adults in the United States have CF.3
What causes cystic fibrosis?
People with CF inherited two copies of a defective or mutated CF gene, meaning each parent was a carrier for CF. Called the cystic fibrosis transmembrane conductance regulator (CFTR) gene, this mutation prevents the CFTR protein from working properly.
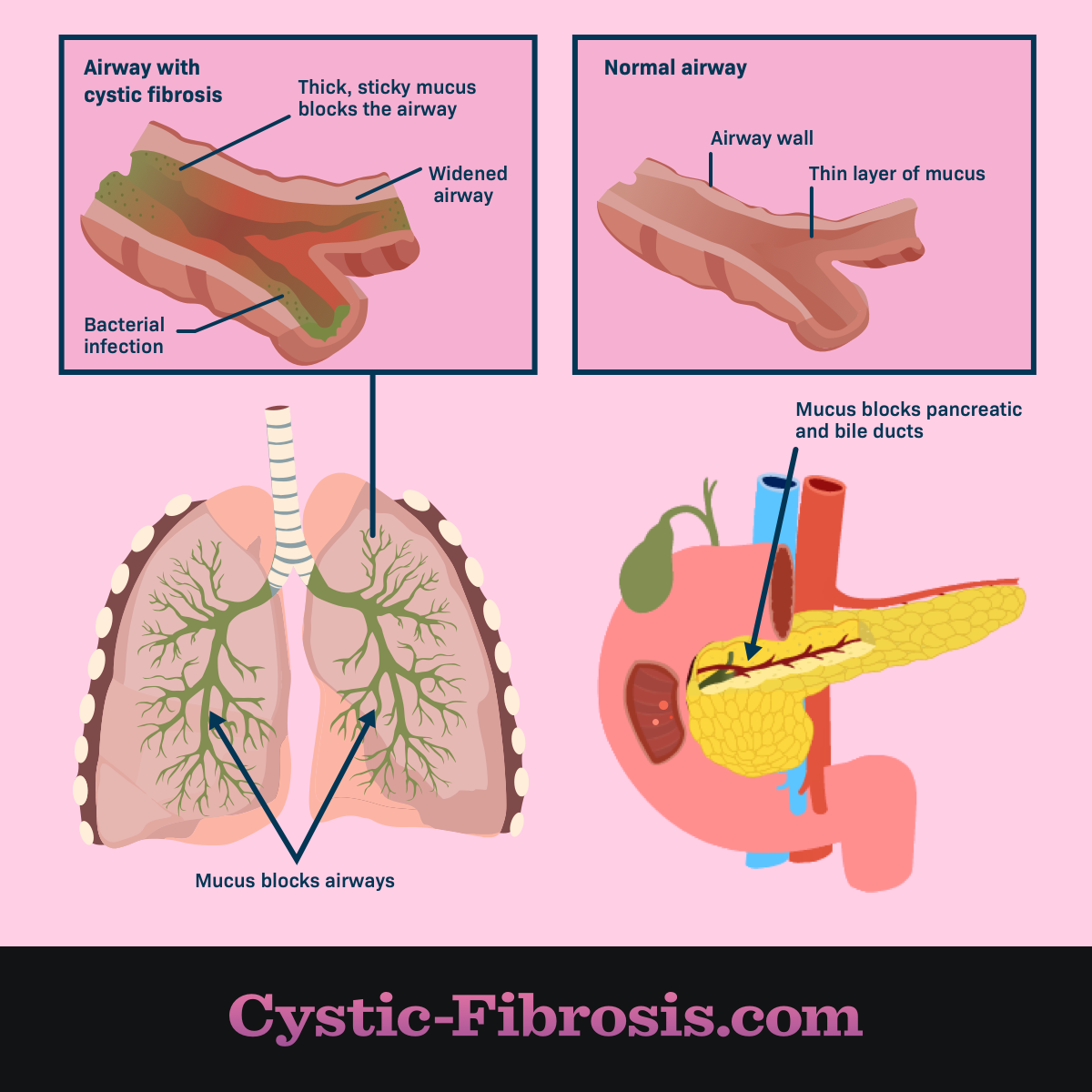
The CFTR protein helps our cells move chloride, an element in salt, to the cell surface. Without chloride, water isn’t attracted to the cell surface, resulting in a thick, sticky mucus building up in the lungs and other organs.
In the lungs, this mucus clogs the airways, which leads to infections, inflammation, and ultimately respiratory failure. In the digestive system, these thick secretions block enzymes that flow from the pancreas to the intestines. Without these enzymes, the body can’t absorb fats and vitamins which causes malnutrition and poor weight gain.1,2
Figure 1. Airway comparisons with and without cystic fibrosis
What are symptoms of cystic fibrosis?
The most common symptoms of cystic fibrosis are:
- Very salty tasting skin (usually noticed when an adult kisses the baby)
- Slow weight gain or failure to thrive even with a good appetite
- Wheezing, coughing, pneumonia
- Greasy, bulky stools or diarrhea
- Nasal polyps
- Chronic sinus infections
- Clubbing or enlargement of the fingertips and toes
Most people with CF are diagnosed soon after birth.1,3
How is cystic fibrosis diagnosed?
In many countries, including the U.S., most babies are screened for CF in the hospital. This newborn screening requires a drop of blood taken during a heel prick. This blood sample screens for several serious illnesses, including cystic fibrosis.
If the test comes back positive for CF, your doctor will conduct a sweat test and genetic tests to confirm or rule out CF. If the results of these tests are inconclusive, more tests may be required. The screening may also mean that your child is a carrier of the faulty CFTR gene but does not have CF.
How is cystic fibrosis treated?
While cystic fibrosis is a complex and serious condition, people with CF are living longer than ever thanks to aggressive research into new treatments and the genetics of CF. The first step in getting treatment is to contact the nearest care center accredited by the Cystic Fibrosis Foundation. These centers specialize in using the latest, research-based evidence to help parents keep their child healthy by avoiding germs, eating smart, and keeping the lungs clear of mucus.
Since the type and severity of CF may vary dramatically from person to person, treatment plans can be very different. That said, some common treatments include:
- Airway clearance techniques
- Inhaled medications
- Antibiotics (to combat infections)
- Pancreatic enzymes
- CFTR modulators
- Nutrition and exercise
The future of cystic fibrosis
The general public generally believes many myths and misconceptions about cystic fibrosis, including that it impacts mostly boys or that people with CF die as children. The fact is that people with CF are living longer, more fulfilling lives thanks to an active support community, expert care, and groundbreaking research into new treatments.